


Published Pictures for 26916 Arf6-GTP mAb
Active Sar1 pulldown assay
Sar1 activation assay kit(NewEast Biosciences, #81801) and the protocol provided with the kit was followed for active Sar1 pulldown from microsomes.
To isolate microsomes, HeLa cells were scraped in ice‐cold PBS and pelleted by centrifugation at 500 g for 10 min. The pellet was resuspended in 400 μl buffer containing 10 mM Hepes‐KOH, pH 7.2, 250 mM sorbitol, 10 mM KOAc, and 1.5 mM MgOAc. Cells were homogenized by passing through a 25‐gauge syringe for at least 30 times and centrifuged at 1,000 g for 10 min. The postnuclear supernatant was then centrifuged at 6,000 g for 10 min. The pellet was resuspended in ice‐cold PBS and centrifuged again at 6,000 g for 10 min. Finally, the pellet was resuspended in 100 μl of 1× assay/lysis buffer provided with the kit and used for active Sar1 pulldown.
To the microsomal fractions, 750 ng of recombinant Rac1 or Sar1 or both, and GTP or GTPγS (100 μM final concentration) or GDP (1 mM final concentration) were added. The reaction mixture was then incubated at 30°C with constant agitation for 30 min for GTP‐loading prior to active Sar1 pulldown with anti‐active Sar1 monoclonal antibody provided in the kit.
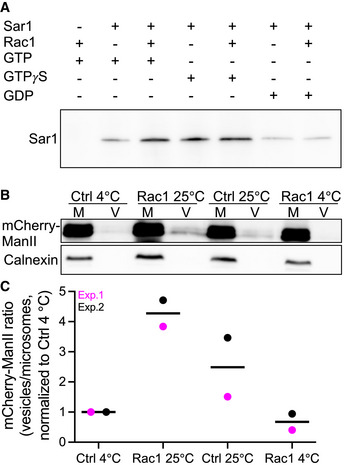
Rac1 stimulates the formation of active Sar1 and COPII
A
Microsomes isolated from HeLa cells were incubated as indicated. Active Sar1 pulldown was then carried out, followed by immunoblotting against Sar1.
B
Microsomes (M) from HeLa cells stably expressing mCherry‐ManII‐RUSH were incubated with ATP and an ATP‐regenerating system, GTP, biotin and cytosol in the presence or absence of recombinant Rac1 for 30 min at 25°C. COPII vesicles (V) were pelleted by ultracentrifugation (100,000 g) and immunoblotted for anti‐mCherry to detect ManII‐RUSH and anticalnexin (ER marker).
C
Quantification data of two independent vesicle budding assays. All values were normalized to 4°C Ctrl condition.
Immunostaining.
Immunostaining was performed as described previously, with minor modifications (51). Briefly, the dissected brains were fixed with PFA (4%) and dehydrated in sucrose (30%), and then sectioned into slices in 40- or 100-μm width. The primary neurons were fixed with PFA (4%) at room temperature or cold methanol at 4 °C for 15 min. The brain slices or primary neurons were first incubated in the blocking buffer (10% FBS, 5% BSA, 0.3% Triton X-100, 0.01% NaN3 dissolved in PBS) at room temperature for 1 h, and then incubated in the primary antibody dissolved in the blocking buffer at 4 °C for overnight, and finally incubated in the secondary antibody conjugated with Alexa Fluor 488, 568, or 647 dyes dissolved in the blocking buffer at room temperature for 1 h. The antibodies used for immunostaining: ….. SAR1-GTP (26916, 1:200; Neweast), ….
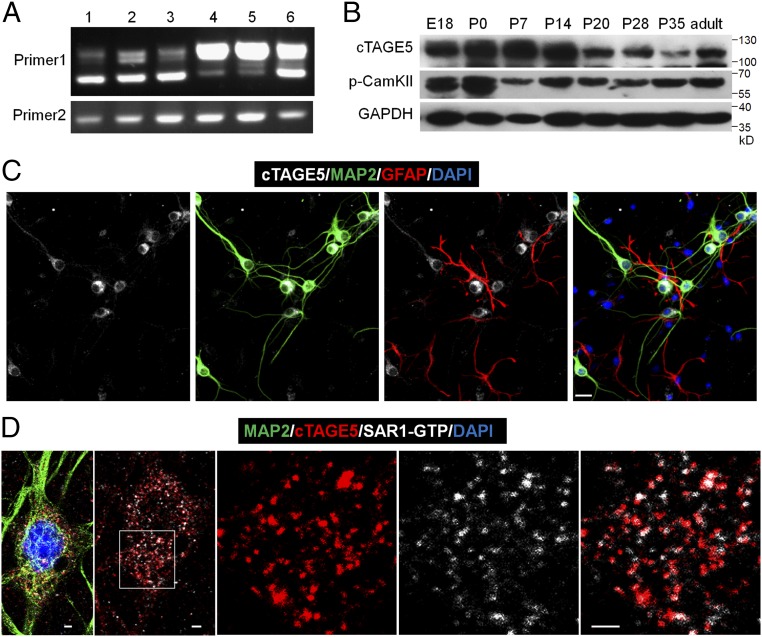
cTAGE5 is expressed in neuron at the ER exit sites.
(A) RT-PCR results showing different splicing isoforms in different tissues during the embryonic stages. The forward primer and reverse primer of “Primer 1” flank the spliced intron of cTAGE5, while “Primer 2” does not. 1: E14 brain; 2: E18 brain; 3: E14 primary neuron; 4: E18 kidney; 5: E18 liver; 6: E14 meninge. (B) Western blotting results showing the expression levels of cTAGE5 and p-CamKII (phospho T286) in the cerebral cortex at different developmental stages. GAPDH was used as loading control. (C and D) Primary neurons (14 div) dissected from E15.5 WT mouse cerebral cortex were stained with cTAGE5 (gray), MAP2 (green), and GFAP (red) antibodies (C); or cTAGE5, SAR1-GTP and MAP2 antibodies (D). (Scale bars, 20 μm in C and 2 μm in D.) Nucleus was stained with DAPI.
Immunostaining.
Immunostaining was performed as described previously, with minor modifications (51). Briefly, the dissected brains were fixed with PFA (4%) and dehydrated in sucrose (30%), and then sectioned into slices in 40- or 100-μm width. The primary neurons were fixed with PFA (4%) at room temperature or cold methanol at 4 °C for 15 min. The brain slices or primary neurons were first incubated in the blocking buffer (10% FBS, 5% BSA, 0.3% Triton X-100, 0.01% NaN3 dissolved in PBS) at room temperature for 1 h, and then incubated in the primary antibody dissolved in the blocking buffer at 4 °C for overnight, and finally incubated in the secondary antibody conjugated with Alexa Fluor 488, 568, or 647 dyes dissolved in the blocking buffer at room temperature for 1 h. The antibodies used for immunostaining: ….. SAR1-GTP (26916, 1:200; Neweast), ….
Co-IP and Western Blotting.
Co-IP and Western blotting were performed as described previously with minor modifications (52). In brief, protein lysates of mouse brain were prepared in the cell lysis buffer [20 mM Tris⋅HCl (pH 8.0), 50 mM NaCl, 10 mM Hepes (pH 7.4), 0.5% Nonidet P-40, 0.5 mM EDTA (pH 8.0), Phosphatase Inhibitor Mixture (Roche), Protease Inhibitor Mixture (Roche), 0.2 mM PMSF]. For Co-IP of endogenous proteins, 2–3 μg of primary antibodies (SAR1-GTP) was first incubated with Protein A/G agarose beads (15 μL + 15 μL) at 4 °C for 2 h, and then the agarose beads were incubated in the 1-mg protein lysates of mouse brain at 4 °C for overnight. After being washed with cell lysis buffer at least three times, the proteins bound on the beads were eluted with SDS sample buffer and analyzed through Western blotting. For Western blotting, the proteins in the brain lysates were separated through SDS/PAGE and transferred into the nitrocellulose (NC) membranes. Then, the NC membranes were incubated in the primary antibodies at 4 °C for overnight and subsequently in the secondary antibodies or heavy-chain specific secondary antibodies (for the co-IP samples) conjugated with horseradish peroxidase. Finally, enhanced chemiluminescence substrate was used to visualize the protein bands in the NC membrane. The antibodies used for Western blotting: …..SAR1-GTP (26916, 1:1,000; Neweast), …
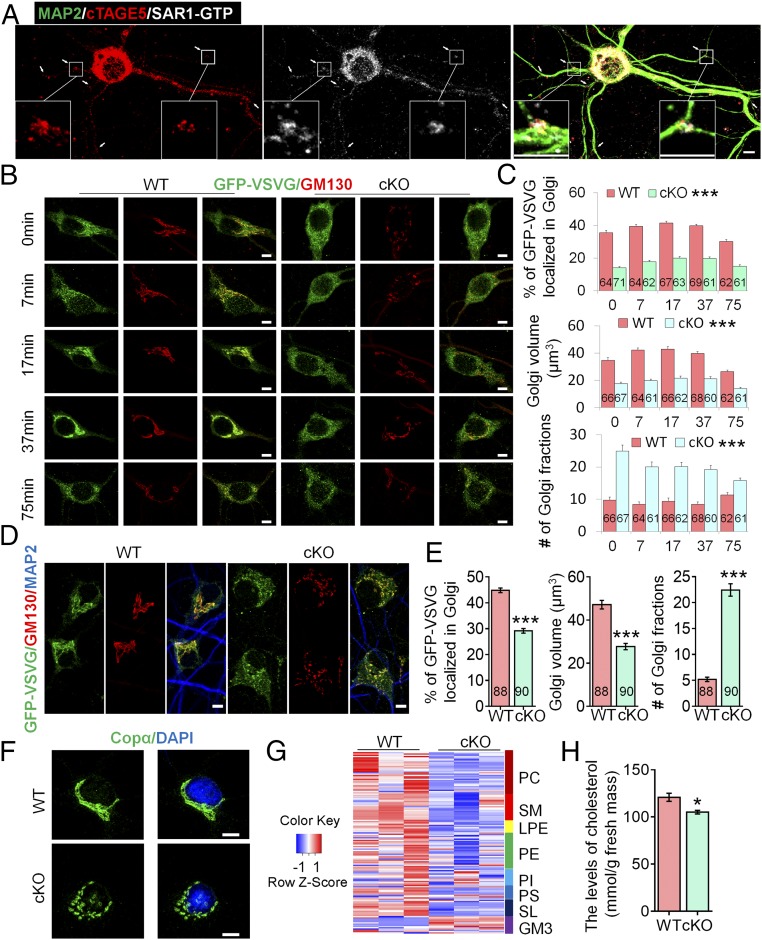
cTAGE5 cKO affects the transport of GFP-VSVG from the ER to the Golgi apparatus in neuron and lipid composition in brain.
(A) Primary neurons at 14 div dissected from around E15.5 WT cerebral cortex were stained with MAP2, cTAGE5, and SAR1-GTP antibodies. Insets are enlarged images of two of the branch points.
Immunostaining.
Immunostaining was performed as described previously, with minor modifications (51). Briefly, the dissected brains were fixed with PFA (4%) and dehydrated in sucrose (30%), and then sectioned into slices in 40- or 100-μm width. The primary neurons were fixed with PFA (4%) at room temperature or cold methanol at 4 °C for 15 min. The brain slices or primary neurons were first incubated in the blocking buffer (10% FBS, 5% BSA, 0.3% Triton X-100, 0.01% NaN3 dissolved in PBS) at room temperature for 1 h, and then incubated in the primary antibody dissolved in the blocking buffer at 4 °C for overnight, and finally incubated in the secondary antibody conjugated with Alexa Fluor 488, 568, or 647 dyes dissolved in the blocking buffer at room temperature for 1 h. The antibodies used for immunostaining: ….. SAR1-GTP (26916, 1:200; Neweast), ….
Co-IP and Western Blotting.
Co-IP and Western blotting were performed as described previously with minor modifications (52). In brief, protein lysates of mouse brain were prepared in the cell lysis buffer [20 mM Tris⋅HCl (pH 8.0), 50 mM NaCl, 10 mM Hepes (pH 7.4), 0.5% Nonidet P-40, 0.5 mM EDTA (pH 8.0), Phosphatase Inhibitor Mixture (Roche), Protease Inhibitor Mixture (Roche), 0.2 mM PMSF]. For Co-IP of endogenous proteins, 2–3 μg of primary antibodies (SAR1-GTP) was first incubated with Protein A/G agarose beads (15 μL + 15 μL) at 4 °C for 2 h, and then the agarose beads were incubated in the 1-mg protein lysates of mouse brain at 4 °C for overnight. After being washed with cell lysis buffer at least three times, the proteins bound on the beads were eluted with SDS sample buffer and analyzed through Western blotting. For Western blotting, the proteins in the brain lysates were separated through SDS/PAGE and transferred into the nitrocellulose (NC) membranes. Then, the NC membranes were incubated in the primary antibodies at 4 °C for overnight and subsequently in the secondary antibodies or heavy-chain specific secondary antibodies (for the co-IP samples) conjugated with horseradish peroxidase. Finally, enhanced chemiluminescence substrate was used to visualize the protein bands in the NC membrane. The antibodies used for Western blotting: …..SAR1-GTP (26916, 1:1,000; Neweast), …
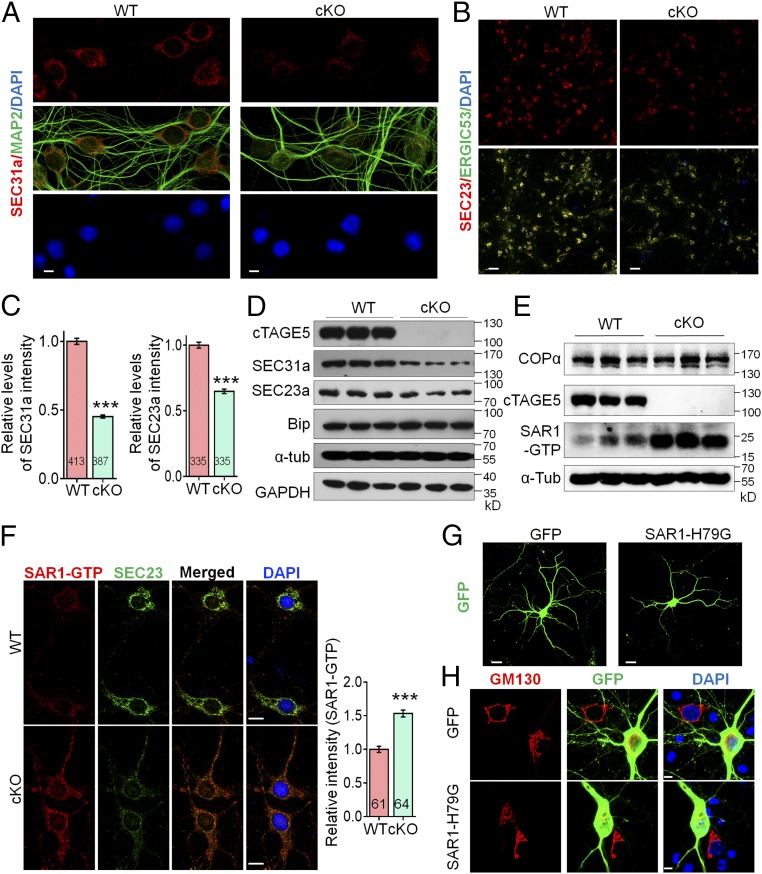
cTAGE5 cKO leads to aberrant expression of COPII components.
(D) or COPα and SAR1-GTP (E) in the cerebral cortex of P20 WT and cKO mice were determined with Western blotting. α-Tub or GAPDH were used as loading control. Three independent replicates were performed. (F) Neurons as those in A and B were stained with SAR1-GTP and SEC23a antibodies. (Right) Quantification of relative staining intensity of SAR1-GTP. t test; ***P < 0.001. Indicated number of neurons from five WT and cKO mice were analyzed. (Scale bars, 10 μm.) (G and H) Primary neurons dissected from around E15.5 WT mouse brain were transfected with SAR1-H79G or GFP cDNA at 8 div, fixed at 14 div, and stained with GFP (G) or GFP and GM130 antibodies (H). (Scale bars, 20 μm in G, 5 μm in H.) Three WT mice were analyzed. The nucleus was stained with DAPI.
Immunostaining.
Immunostaining was performed as described previously, with minor modifications (51). Briefly, the dissected brains were fixed with PFA (4%) and dehydrated in sucrose (30%), and then sectioned into slices in 40- or 100-μm width. The primary neurons were fixed with PFA (4%) at room temperature or cold methanol at 4 °C for 15 min. The brain slices or primary neurons were first incubated in the blocking buffer (10% FBS, 5% BSA, 0.3% Triton X-100, 0.01% NaN3 dissolved in PBS) at room temperature for 1 h, and then incubated in the primary antibody dissolved in the blocking buffer at 4 °C for overnight, and finally incubated in the secondary antibody conjugated with Alexa Fluor 488, 568, or 647 dyes dissolved in the blocking buffer at room temperature for 1 h. The antibodies used for immunostaining: ….. SAR1-GTP (26916, 1:200; Neweast), ….
Co-IP and Western Blotting.
Co-IP and Western blotting were performed as described previously with minor modifications (52). In brief, protein lysates of mouse brain were prepared in the cell lysis buffer [20 mM Tris⋅HCl (pH 8.0), 50 mM NaCl, 10 mM Hepes (pH 7.4), 0.5% Nonidet P-40, 0.5 mM EDTA (pH 8.0), Phosphatase Inhibitor Mixture (Roche), Protease Inhibitor Mixture (Roche), 0.2 mM PMSF]. For Co-IP of endogenous proteins, 2–3 μg of primary antibodies (SAR1-GTP) was first incubated with Protein A/G agarose beads (15 μL + 15 μL) at 4 °C for 2 h, and then the agarose beads were incubated in the 1-mg protein lysates of mouse brain at 4 °C for overnight. After being washed with cell lysis buffer at least three times, the proteins bound on the beads were eluted with SDS sample buffer and analyzed through Western blotting. For Western blotting, the proteins in the brain lysates were separated through SDS/PAGE and transferred into the nitrocellulose (NC) membranes. Then, the NC membranes were incubated in the primary antibodies at 4 °C for overnight and subsequently in the secondary antibodies or heavy-chain specific secondary antibodies (for the co-IP samples) conjugated with horseradish peroxidase. Finally, enhanced chemiluminescence substrate was used to visualize the protein bands in the NC membrane. The antibodies used for Western blotting: …..SAR1-GTP (26916, 1:1,000; Neweast), …
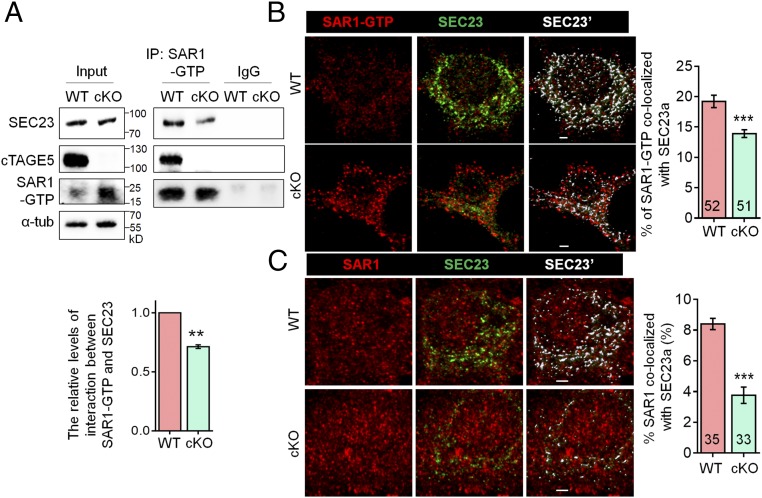
cTAGE5 cKO disturbs the interaction between SEC23 and SAR1.
(A) Tissue lysates of P20 WT and cKO mouse cortex were immunoprecipitated with SAR1-GTP antibody and probed with SEC23, cTAGE5, and SAR1-GTP antibodies. The relative levels of interaction between SAR1-GTP and SEC23 was defined as the level of SEC23 in the immunocomplex, normalized with the level of SEC23 in the input and SAR1-GTP in immunocomplex. Paired t test; **P < 0.01. n = 3. (B and C) Representative images of primary neurons at 14 div, which were dissected from around E15.5 WT and cKO mouse brains and stained with SAR1-GTP (B) or SAR1 (C) and SEC23 antibodies. (Right) Quantification of SAR1-GTP (B) or SAR1 (C) colocalized with SEC23. The “Surface” function of Imaris software was used to simulate the distribution of SEC23 (white SEC23′), and the portion of SAR1-GTP or SAR1 localized on the SEC23 surface was determined as colocalized with SEC23. t test; ***P < 0.001. Indicated number of neurons from five (for SAR1-GTP) and four (for SAR1) WT and cKO mice were analyzed. (Scale bars, 2 μm.)
Immunocytochemistry
Primary cortical neurons were transfected with eGFP or HIP14-GFP or APT1-turboGFP plasmid at DIV0, infected at DIV3 with BDNF-mCherry, and then fixed at DIV10 with 4% PFA 20 min at room temperature and then, after one wash in PBS, treated for 15 min with 50 mM NH4Cl. After permeabilization with 0.1% Triton X-100, neurons were incubated for 1 hour at room temperature with blocking solution (0.1% Triton X-100 and 5% NGS in PBS). The incubation with primary antibodies was made overnight at 4°C in blocking solution: …. anti-Active Sar1-GTP (Biomol-NewEast Biosciences #NB-26916; 1:100), …. After 2 hours of incubation at room temperature with II antibodies, the coverslips were incubated with Hoechst (Sigma-Aldrich #B2261; 1:5000) and mounted on glass slides with ProLong Diamond Antifade Mounting (Life Technologies #P36961). Images were acquired with a 63× oil-immersion objective using an inverted confocal microscope (LSM 710, Zeiss) coupled to an Airyscan detector.
For immunofluorescence on mouse Thy1-p50 GFP brain slices, samples were fixed after mouse perfusion for 2 hours in 4% PFA, washed three times with PBS, and then incubated for 16 hours in 20% sucrose in PBS. Brains were then incubated in 30% sucrose in PBS. After inclusion in Tissue-Tek Optimal Cutting Temperature (OCT), samples were frozen at −20°C and kept at −80°C for long storage. Slices (35 μm) were prepared at the cryostat and mounted on Thermo Fisher Scientific SuperFrost Ultra Plus GOLD Adhesion Slides. At this step, slices were acquired with a 20× objective (0.45 NA) using a slide scanner (AxioScan Z1, Zeiss) or processed for immunostaining as follows. An unmasking step was performed with Citrate buffer (Sigma-Aldrich #C9999; 1:1000 in H2O) for 20 min at 95°C and then slices were incubated with 5% Normal Donkey Serum in PBS–0.3% Triton X-100 for 1 hour. Primary antibodies were incubated overnight at 4°C in blocking solution with the following concentrations: anti-CTIP2 (Abcam #Ab18465; 1:300), anti-CUX1 (Novus Biologicals #NBP2-13883; 1:100), anti-TBR1 (Abcam #ab31940; 1:200), anti-GFP (Millipore #AB16901; 1:500), anti-GRASP65 (Thermo Fisher Scientific #PA3-910; 1:300), and anti-calnexin (Sigma-Aldrich #C4731; 1:500). After incubation with secondary antibodies, samples were incubated with Hoechst (Sigma-Aldrich #B2261; 1:5000) and then coverslips were mounted with Dako Fluorescent Mounting Medium (Agilent). Acquisitions were made using a 20× or 40× oil-immersion objective using an inverted confocal microscope (LSM 710, Zeiss) and z-stacks were made with five slices of 5 μm each for a total of 20 μm. For calnexin and GRASP65 staining, images were acquired with a 63× oil-immersion objective (1.4 NA) using an inverted confocal microscope (LSM 710, Zeiss) coupled to an Airyscan detector to improve signal-to-noise ratio and increase resolution; z-stacks were made with 20 to 25 slices of 0.3 μm each for a total of 6 to 7 μm.
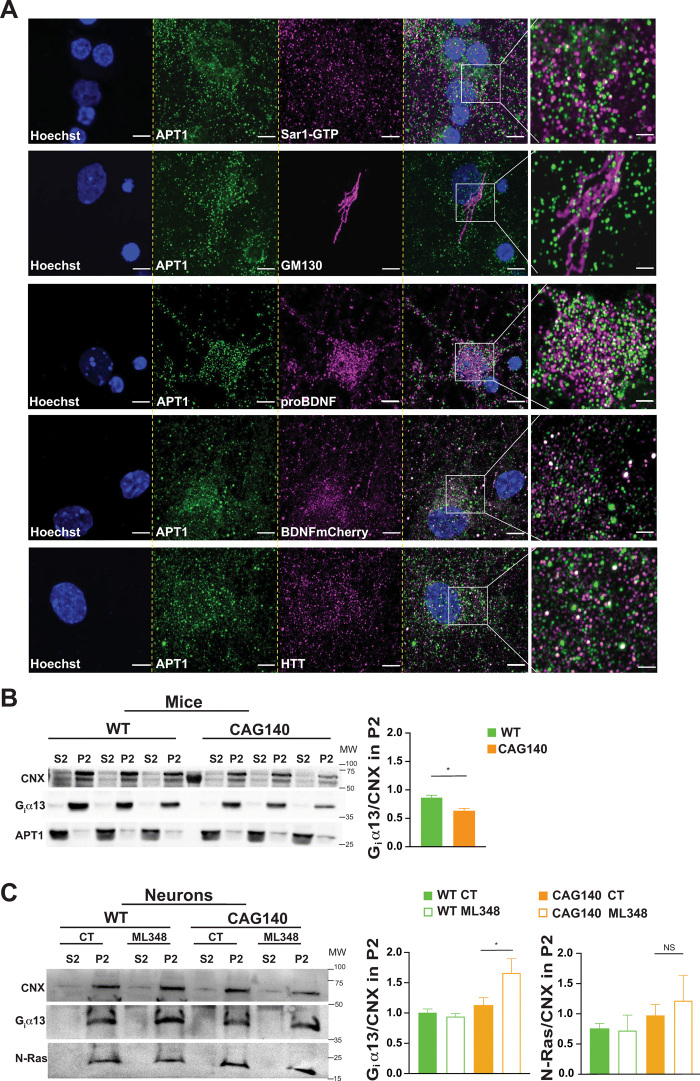
APT1 is expressed in the neuronal secretory pathway.
(A) Confocal images of endogenous APT1 in cortical neurons showing colocalization of the enzyme with proteins related to COPII (Sar1-GTP), Golgi apparatus (GM130), secreted dense core vesicles (proBDNF and BDNF-mCherry), and HTT (4C8 mAb). Nuclei were counterstained with Hoechst (scale bars, 5 μm). For each staining, the images were zoomed five times in a specific region of interest and processed by an Airyscan detector (inset scale bar, 2 μm).